And the evidence keeps rolling in:-) At this pace, I may be able to retire sooner (or later, hopefully sooner). Here's another recent LinkedIn post I authored explaining that a drug that was originally developed to treat cancer was successfully used to improve symptoms in children with Progeria. Interestingly, that same drug was also recently shown by researchers at the University of North Carolina and Case Western Reserve University (in collaboration with the drug company Merck) to reactivate latent HIV-1 in T cells taken directly from patients infected with HIV-1. One big coincidence?? I think not. This is just another example of researchers not realizing that many chemically distinct compounds converge on and indirectly activate a common pathway (AMPK) that connects and characterizes nearly every cell, system, and ultimately disease pathology within the body. Is it possible that normal aging, accelerated aging, infections that involve dormant viruses (e.g. HIV-1, HPV, Herpes, etc), and cancer are all connected and can be treated or possibly eradicated by activating a particular pathway that increases the lifespan and healthspan of nearly every organism that it's been tested in? If so, I think my mom will be pretty proud of me (if she actually believes me:-)
Cancer drug used to successfully treat children with Progeria also reactivates latent HIV-1: Connection between AMPK, aging, and HIV-1 reactivation
In an abstract presented in December of 2013 at the Sixth International Workshop on HIV Persistence during Therapy, researchers from the University of North Carolina and Case Western Reserve University (in collaboration with the pharmaceutical company Merck) reported that the use of an ultra-high throughput screen identified a class of compounds known as farnesyl-transferase inhibitors (FTIs) that reactivated latent HIV-1 expression and acted synergistically with other agents to reverse latency in T cell models and in resting CD4+ T cells isolated from patients infected with HIV-1 [1].
Interestingly, of the sub-classes of FTI’s identified in the screen that synergistically reactivated latent HIV-1, the FTI lonafarnib was specifically listed as a representative structure identified in the screen [1]. The identification of lonafarnib as a HIV-1 latency reversing agent is startling considering that lonafarnib, originally tested in preclinical studies as an anti-cancer agent, has recently been shown and reported to be beneficial in a clinical trial for the treatment of children diagnosed with Hutchinson-Gilford progeria syndrome (HGPS) [2,3].
The mechanism of action of FTI’s involves the inhibition of an enzyme known as farnesyl-transferase (FT). FT is an enzyme that adds a farnesyl group to proteins that bear a particular amino acid sequence [4]. The farnesyl group typically targets the protein to the cell membrane, often facilitating cellular signaling involving the farnesylated protein [4]. Within the context of HGPS, the protein lamin A becomes permanently farnesylated (also known as progerin) due to a gene mutation, leading to an excessive accumulation of progerin in the nuclear membrane and thus generating nuclear blebbing and dysfunctional nuclear processes [5]. Similarly, the oncoprotein Ras, a protein critical for cell cycle progression that is abnormally active in many cancers, is farnesylated by FT, thereby facilitating attachment of Ras to the cell membrane to promote signal transduction [4].
Although the primary mechanism of action of lonafarnib is the inhibition of FT, it is likely, similar to rapamycin and other compounds that are effective in treating progeria but also have immuno-modulatory effects, that inhibition of FT by lonafarnib is generating an indirect activation of the master metabolic regulator AMPK via the induction of cellular stress (i.e. anabolic pathway inhibition). Indeed, FT is also involved in the mevalonate pathway, an anabolic pathway that is characterized by the biosynthesis of cholesterol [6]. Interestingly, statins, which function by inhibiting the rate limiting enzyme in the mevalonate pathway, HMG-CoA reductase, activate AMPK and have been shown to be efficacious in preclinical animal models of progeria [7,8].
In the abstract presented at the Sixth International Workshop on HIV Persistence during Therapy, the researchers noted that FTI’s act synergistically with the latency reversing agent JQ1 and protein kinase C (PKC) activators [1]. Interestingly, as AMPK activation is essential for T cell activation and thus efficient latent HIV-1 reactivation, JQ1 as well as the PKC activator bryostatin have both been shown to induce the phosphorylation and activation of AMPK in recent studies, implying that the FTI lonafarnib likely also induces AMPK activation [9,10].
Indeed, the abstract presented intriguing data that knock down of the beta subunit of the FT enzyme sensitized and constitutively reactivated latent HIV-1 in a Jurkat T cell model system [1]. Such evidence is provocative in the sense that the inhibition or knock down of mediators that function to promote anabolic processes, including HMG-CoA reductase (cholesterol biosynthesis), ATIC/AICART (purine metabolism), and mTOR (protein synthesis), lead to cellular stress-induced activation of AMPK [7,11,12]. Similarly, the inhibition of the beta subunit of FT, which is also involved in the anabolic mevalonate pathway, likely induces a cellular stress-induced activation of AMPK, potentially explaining a portion of the beneficial effects of lonafarnib in both HGPS and latent HIV-1 reactivation.
Lastly, the abstract also presented evidence that FTIs acted synergistically with the HDAC inhibitor vorinostat to reactivate latent HIV-1 in resting CD4+ memory T cells isolated from HIV-1 patients [1]. Interestingly, vorinostat has also been shown to induce AMPK activation in certain cancer cells [13].
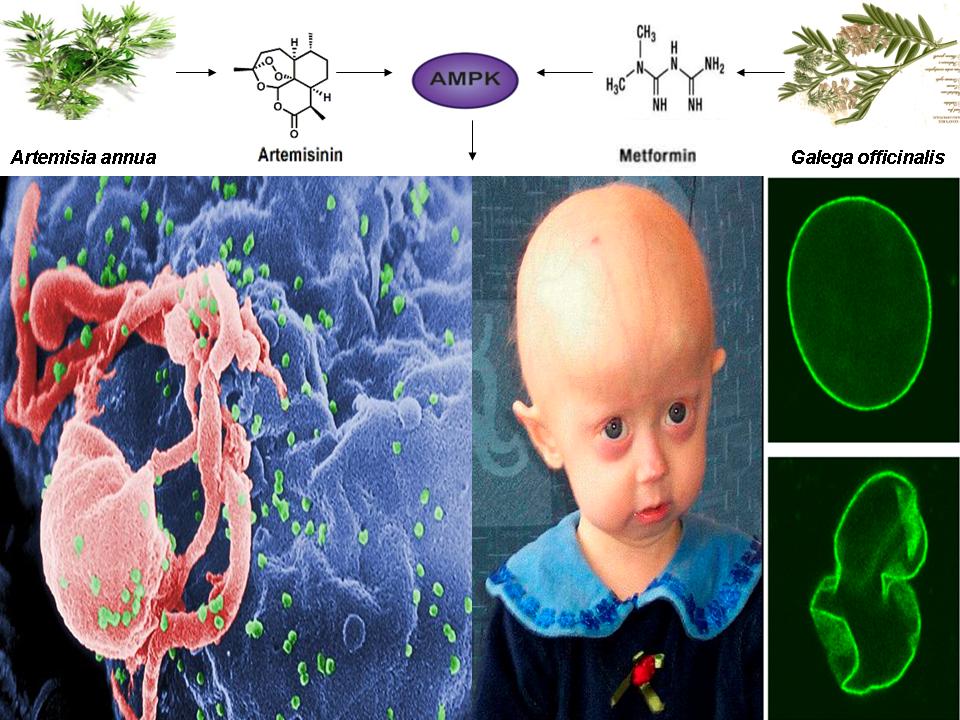
Collectively, data from this abstract and the recent results published from the HGPS clinical trial provides additional evidence (reinforcing the implications of the figure below) that inhibition of FT by FTI’s such as lonafarnib leads to the induction of AMPK activation via the promotion of a cellular stress response, not unlike that of mTOR inhibition-induced AMPK activation by rapamycin [12]. The novel observation that chemically distinct compounds converge on single pathway to effectuate beneficial results in diseases as seemingly disparate as HGPS and HIV-1 reactivation is unprecedented.

https://www.linkedin.com/pulse/cancer-drug-used-successfully-treat-children-progeria-finley
References:
- Farnesyl-transferase inhibitors: identification and validation of a class which reactivates HIV latent expression and is synergistic with other mechanisms in vitro. In: 6th intl workshop on HIV persistance during therapy report summary – report by David Margolis MD, UNC chapel hill and the collaboratory of AIDS researchers for eradication (CARE) – (12/15/13). http://www.natap.org/2013/HIV/121913_01.htm, last accessed 04/24/2016.
- Morgillo F, Lee HY. Lonafarnib in cancer therapy. Expert Opin Investig Drugs. 2006 Jun;15(6):709-19.
- Gordon LB, Kleinman ME, Miller DT, et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2012 Oct 9;109(41):16666-71.
- Reuter CW, Morgan MA, Bergmann L. Targeting the Ras signaling pathway: a rational, mechanism-based treatment for hematologic malignancies? Blood. 2000 Sep 1;96(5):1655-69.
- Prokocimer M, Barkan R, Gruenbaum Y. Hutchinson-Gilford progeria syndrome through the lens of transcription. Aging Cell. 2013 Aug;12(4):533-43.
- Marcuzzi A, De Leo L, Decorti G, Crovella S, Tommasini A, Pontillo A. The farnesyltransferase inhibitors tipifarnib and lonafarnib inhibit cytokines secretion in a cellular model of mevalonate kinase deficiency. Pediatr Res. 2011 Jul;70(1):78-82.
- Sun W, Lee TS, Zhu M, et al. Statins activate AMP-activated protein kinase in vitro and in vivo. Circulation. 2006 Dec 12;114(24):2655-62.
- Varela I, Pereira S, Ugalde AP, et al. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat Med. 2008 Jul;14(7):767-72.
- Wang H, Sharma L, Lu J, Finch P, Fletcher S, Prochownik EV. Structurally diverse c-Myc inhibitors share a common mechanism of action involving ATP depletion. Oncotarget. 2015 Jun 30;6(18):15857-70.
- Mehla R, Bivalkar-Mehla S, Zhang R, et al. Bryostatin modulates latent HIV-1 infection via PKC and AMPK signaling but inhibits acute infection in a receptor independent manner. PLoS One. 2010 Jun 16;5(6):e11160.
- Asby DJ, Cuda F, Beyaert M, Houghton FD, Cagampang FR, Tavassoli A. AMPK Activation via Modulation of De Novo Purine Biosynthesis with an Inhibitor of ATIC Homodimerization. Chem Biol. 2015 Jul 23;22(7):838-48.
- Chiao YA, Kolwicz SC, Basisty N, et al. Rapamycin transiently induces mitochondrial remodeling to reprogram energy metabolism in old hearts. Aging (Albany NY). 2016 Feb;8(2):314-27.
- Sarfstein R, Bruchim I, Fishman A, Werner H. The mechanism of
action of the histone deacetylase inhibitor vorinostat involves
interaction with the insulin-like growth factor signaling pathway. PLoS
One. 2011;6(9):e24468.