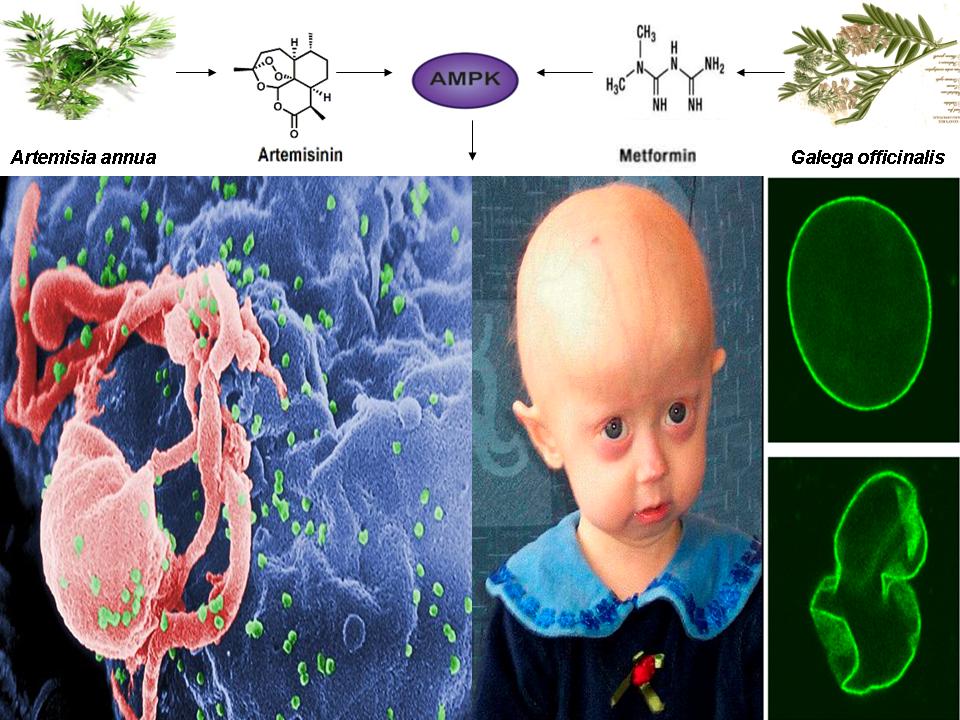
Another recent LinkedIn post I authored (see below) showing that a naturally occurring compound that is used as a powerful first line treatment for malaria activates the same pathway as the broccoli sprout compound sulforaphane, the vitamin A metabolite retinoic acid, metformin derived from the French Lilac, and methylene blue. This compound, called artemisinin, won the 2015 Nobel prize in Medicine for a Chinese scientist and has been used for over 2000 years to treat malaria dating back to 200 BC. This same compound has been shown to inhibit cancer by activating AMPK and mimic calorie restriction by increasing energy levels (ATP) and mitochondrial biogenesis. Just as sulforaphane, retinoic acid, and methylene blue also inhibit cancer and malaria and reverse aging in progeria cells, artemisinin (derived from the plant Artemisia annua) will likely also reverse symptoms of accelerated aging in progeria cells and in normal humans and reactivate latent HIV-1. Again, if further evidence proves this to be true............:-)
Nobel Prize Winning drug Artemisinin shares common mechanism of action with AMPK activator Metformin: Connection between aging and HIV-1 reactivation
A recent study published online in the journal Oncotarget in June of 2015 provided incredible evidence that a common metabolite of artemisinin and its analogs, a powerful anti-malarial drug derived from the plant Artemisia annua (a plant that has been used by Chinese herbalists for more than 2000 years to treat malaria, with the earliest recording dating back to 200 BC) shares a common mechanism of action with several other chemically distinct compounds (including a compound that has shown powerful effects in reactivating latent HIV-1) that is dependent on the activation of the master metabolic regulator AMPK [1,2].
Artemisinin or its semi-synthetic derivatives (which are all metabolized into dihydroartemisinin) are considered the standard first-line treatment for malaria caused by the protozoan parasite Plasmodium falciparum [3]. Half of the 2015 Nobel Prize in Medicine was awarded to the Chinese scientist Tu Youyou for her discovery of artemisinin [4]. Although the mechanism of action of artemisinin has been vigorously debated for decades and is currently unknown, the generation of free radicals leading to death of the parasite is a widely-accepted theory [5].
In addition to its anti-malarial effects, dihydroartemisinin has also been shown to exert potent anti-cancer effects in a range of malignancies. In cancer research, artemisinin is characterized as a synthetic lethal c-Myc inhibitor [1]. c-Myc is one of the most frequently deregulated oncoproteins (a protein that could contribute to oncogenesis due to mutations or increased expression) in human cancers and is also upregulated during HIV-1 latency [6,7]. Interestingly, in the Oncotarget study, the authors demonstrated that regardless of the class to which each studied drug had been placed (i.e., direct, indirect, or synthetic lethal c-Myc inhibitors), the therapeutic action of each drug converges on a single overriding mechanism that involves the activation of AMPK due to depletion of ATP stores induced by mitochondrial dysfunction, resulting in cell cycle arrest and differentiation or apoptosis (i.e. cell death) of several cancer cell lines [1].
Although structurally dissimilar and considered an indirect c-Myc inhibitor in cancer research, the BRD4 inhibitor JQ1 was also shown to act via the same mechanism of action as dihydroartemisinin in this study via induction of mitochondrial dysfunction, reduced ATP levels, activation of AMPK, and cell cycle arrest and differentiation of leukemia cells [1].
The authors also reasoned that if a common mechanism of action of mitochondrial dysfunction and compensatory AMPK activation links dihydroartemisinin and JQ1, then the use of well-characterized mitochondrial inhibitors that also activate AMPK should produce the same or similar results as dihydroartemisinin and JQ1. Indeed, the use of metformin (inhibits complex I of the mitochondrial electron transport chain) or oligomycin (inhibits complex V of the mitochondrial electron transport chain) also lead to a decrease in Myc protein levels, reduction in the levels of ATP, activation of AMPK, and cancer cell differentiation, providing convincing evidence that compounds as chemically distinct as metformin, dihydroartemisinin, JQ1, and oligomycin share a common mechanism of action that centers on AMPK activation [1]. Moreover, artemisinin has been shown in another recent study to inhibit neuroblastoma proliferation (using SH-SY5Y cells) via the activation of AMPK, as AMPK inhibition by compound C or siRNA abrogated artemisinin’s effects [8].
Interestingly, also in the Oncotarget study, the use of the phorbol ester TPA/PMA, produced similar results as metformin, dihydroartemisinin, and JQ1 on cancer cell differentiation [1]. As noted above, JQ1 has also been shown to be a powerful re-activator of latent HIV-1, facilitating its eventual detection and destruction by the immune system, and PMA combined with ionomycin (a chemical that increases the levels of calcium in a cell) is such a powerful reactivator of latent HIV-1 in T cells that it is used as a benchmark positive control in latent HIV-1 reactivation studies [9,10].
Bryostatin-1, a protein kinase C modulator, was shown to activate AMPK and, when combined with metformin, reactivate latent HIV-1 in the THP-p89 monocytic cell line in an AMPK-dependent manner [11]. As AMPK is essential for T cell activation, bryostatin-1 combined with JQ-1 increased surface expression of T cell activation markers that reactivate latent HIV-1 to levels comparable with the levels of positive controls [12]. Furthermore, the phorbol ester PMA was also shown to induce differentiation of SH-SY5Y human neuroblastoma cells (similar to retinoic acid and dihydroartemisinin) in an AMPK-dependent manner [1,13,14]. Given that c-Myc (which is inhibited by metformin and dihydroartemisinin) is upregulated during HIV-1 latency and directly activates transcription of the splicing factor SRSF1 (a splicing factor that is deregulated in Hutchinson-Gilford progeria syndrome and also promotes HIV-1 latency), a common mechanism of action potentially links HIV-1 latency and progeria with chemically distinct compounds [7,15].
More remarkable, however, is a study recently published online in the journal PeerJ in March of 2015 demonstrating that artesunate (in doses much lower than those used to treat malaria) mimics certain aspects of caloric restriction when given to mice, triggers mitochondrial biogenesis, and attenuates telomere attrition [16]. Administration of artesunate, a semi-synthetic derivative of artemisinin that is metabolized to dihydroartemisinin, to mice lead to an increase in endothelial nitric oxide synthase (eNOS) with an accompanying increase in the levels of nitric oxide and COX4 (a component of the mitochondrial respiratory chain) that was mirrored by the application of hydrogen peroxide (H2O2), indicating that an oxidative burst is responsible for enhancement of mitochondrial structure and function by artesunate [16].
Indeed, skeletal muscle cells from artesunate treated mice possessed more mitochondrial layers than control cells and the levels of mitochondrial localized SIRT3, the antioxidant enzymes Mn-SOD (primarily localized to the mitochondria), Cu-ZN/SOD, and catalase, as well as glutathione were upregulated in response to both artesunate and H2O2, indicative again of increased mitochondrial functionality [16]. Most importantly, however, artesunate increased the phosphorylation and activation of both AMPK and eNOS (which is activated by AMPK), upregulated SIRT1 levels (increases life- and healthspan), increased PGC1-alpha levels (master transcriptional regulator of mitochondrial biogenesis), and increased the levels of the mitochondrial biomarkers CYT C and MNF2. Increased telomere length was also observed compared to control mice [16].
Collectively, the Oncotarget and PeerJ studies paint an astonishing molecular portrait in which artemisinin’s mechanism of action likely converges with that of metformin, JQ1, bryostatin-1, oligomycin, and many others via a single overriding mechanism that is orchestrated by stress-induced (e.g. free radical generation, increase in intracellular calcium levels, increase in the AMP/ATP ratio) activation of AMPK. Indeed, compounds that generate oxidative stress (H2O2, etc.) are well-characterized activators of AMPK and have also been shown to reactivate latent HIV-1 [17,18].
Enhanced mitochondrial functionality is also essential for T cell activation/latent HIV-1 reactivation as well as in the amelioration or correction of accelerated aging defects in progeria [19,20]. Interestingly, just as artemisinin has been shown to activate AMPK, increase PGC-1 alpha levels, and enhance mitochondrial biogenesis and functionality, methylene blue has also been shown to activate AMPK and correct nuclear morphology and reverse accelerated aging defects in progeria cells by increasing PGC-1a levels and enhancing mitochondrial functionality, implying that artemisinin may also act to slow or reverse aging defects in progeria cells [19,21].
Additionally, all-trans retinoic/retinoic acid signaling also activates AMPK, is essential for T cell activation, promotes the differentiation of SH-SY5Y cells similar to PMA (artemisinin also inhibits SH-SY5Y proliferation), reduces the levels of the splicing factor SRSF1 (a splicing factor that is activated by c-Myc, upregulated in HIV-1 latency, and deregulated in progeria), and decreases progerin mRNA levels in progeria cells, leading to amelioration of accelerated aging defects [see prior post on retinoic acid for references]. Also, the broccoli sprout compound sulforaphane has been shown to activate AMPK, stimulate the immune response, decrease epigenetic factors that promote HIV-1 latency, and reverse accelerated aging symptoms in progeria cells by inducing autophagy of the mutant progerin protein [see prior post on sulforaphane for references]. Interestingly, AMPK activation is essential for the induction of autophagy and artemisinin activates AMPK and upregulates autophagy, giving further indication that artemisinin may correct accelerating aging defects in progeria cells, reactivate latent HIV-1, and potentially ameliorate symptoms of aging in normal humans as well [22,23].
Interestingly, artesunate has been shown to increase the suicidal death of erythrocytes (i.e. eryptosis) via modulation of intracellular calcium or oxidative stress, which may potentially accelerate elimination of infected erythrocytes (i.e. red blood cells) prior to exit by Plasmodium falciparium, thus reducing parasitemia and positively influencing the course of malaria. Surprisingly, in addition to an increase in intracellular calcium or oxidative stress, both of which activate AMPK, several naturally-occurring compounds that are known activators of AMPK also induce suicidal death in erythrocytes, including curcumin (from the spice turmeric), ursolic acid (found in many fruits and herbs including apples, basil, bilberries, etc.), retinoic acid, and sulforaphane [27-29].
Lastly, as an example of the existence of a common mechanism of AMPK activation among distinct compounds, just as artemisinin is not only a powerful anti-malarial drug but also activates AMPK, PGC-1a, Sirtuins, and inhibits telomere attrition, methylene blue, sulforaphane, and retinoic acid have each been shown to have additional anti-malarial effects as evidenced by phagocytosis (i.e. cell “eating” or engulfment) of an erythrocyte infected with Plasmodium falciparium, providing additional indications that artemisinin may prove efficacious in the treatment of progeria and the reversal of HIV-1 latency [24-26]. If further evidence substantiates this interconnectedness, a paradigm shift in the treatment and prevention of diseases once thought incurable will be inevitable.
https://www.linkedin.com/pulse/nobel-prize-winning-drug-artemisinin-shares-common-mechanism-finley?trk=mp-reader-card
References:
1. Wang H, Sharma L, Lu J, Finch P, Fletcher S, Prochownik EV. Structurally diverse c-Myc inhibitors share a common mechanism of action involving ATP depletion. Oncotarget. 2015 Jun 30;6(18):15857-70.
2. http://wwwnc.cdc.gov/eid/article/20/7/ET-2007_article
3. Woodrow CJ, Haynes RK, Krishna S. Artemisinins. Postgrad Med J. 2005 Feb;81(952):71-8.
4. http://www.nobelprize.org/nobel_prizes/medicine/laureates/2015/press.pdf
5. Meshnick SR. Artemisinin: mechanisms of action, resistance and toxicity. Int J Parasitol. 2002 Dec 4;32(13):1655-60.
6. Morton JP, Sansom OJ. MYC-y mice: from tumour initiation to therapeutic targeting of endogenous MYC. Mol Oncol. 2013 Apr;7(2):248-58
7. Jiang G, Espeseth A, Hazuda DJ, Margolis DM. C-Myc and Sp1 contribute to proviral latency by recruiting histone deacetylase 1 to the human immunodeficiency virus type 1 promoter. J Virol 2007;81(20):10914–23.
8. Tan WQ, Chen G, Jia B, Ye M. Artemisinin inhibits neuroblastoma proliferation through activation of AHP-activated protein kinase (AMPK) signaling. Pharmazie. 2014 Jun;69(6):468-72.
9. Darcis G, Kula A, Bouchat S, et al. An In-Depth Comparison of Latency-Reversing Agent Combinations in Various In Vitro and Ex Vivo HIV-1 Latency Models Identified Bryostatin-1+JQ1 and Ingenol-B+JQ1 to Potently Reactivate Viral Gene Expression. PLoS Pathog. 2015 Jul 30;11(7):e1005063.
10. Spina CA, Anderson J, Archin NM, et al. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathog. 2013;9(12):e1003834.
11. Mehla R, Bivalkar-Mehla S, Zhang R, et al. Bryostatin modulates latent HIV-1 infection via PKC and AMPK signaling but inhibits acute infection in a receptor independent manner. PLoS One. 2010 Jun 16;5(6):e11160.
12. Laird GM, Bullen CK, Rosenbloom DI, et al. Ex vivo analysis identifies effective HIV-1 latency–reversing drug combinations. J Clin Invest. 2015 May;125(5):1901-12.
13. Zogovic N, Tovilovic-Kovacevic G, Misirkic-Marjanovic M, et al. Coordinated activation of AMP-activated protein kinase, extracellular signal-regulated kinase, and autophagy regulates phorbol myristate acetate-induced differentiation of SH-SY5Y neuroblastoma cells. J Neurochem. 2015 Apr;133(2):223-32.
14. Meseguer S, Mudduluru G, Escamilla JM, Allgayer H, Barettino D. MicroRNAs-10a and -10b contribute to retinoic acid-induced differentiation of neuroblastoma cells and target the alternative splicing regulatory factor SFRS1 (SF2/ASF). J Biol Chem. 2011 Feb 11;286(6):4150-64.
15. Das S, Anczuków O, Akerman M, Krainer AR. Oncogenic splicing factor SRSF1 is a critical transcriptional target of MYC. Cell Rep 2012;1(2):110–7.
16. Wang DT, He J, Wu M, Li SM, Gao Q, Zeng QP. Artemisinin mimics calorie restriction to trigger mitochondrial biogenesis and compromise telomere shortening in mice. PeerJ. 2015 Mar 5;3:e822.
17. Auciello FR, Ross FA, Ikematsu N, Hardie DG. Oxidative stress activates AMPK in cultured cells primarily by increasing cellular AMP and/or ADP. FEBS Lett. 2014 Sep 17;588(18):3361-6.
18. Piette J, Legrand-Poels S. HIV-1 reactivation after an oxidative stress mediated by different reactive oxygen species. Chem Biol Interact. 1994 Jun;91(2-3):79-89.
19. Xiong ZM, Choi JY, Wang K, et al. Methylene blue alleviates nuclear and mitochondrial abnormalities in progeria. Aging Cell. 2015 Dec 14. doi: 10.1111/acel.12434.
20. Ron-Harel N, Sharpe AH, Haigis MC. Mitochondrial metabolism in T cell activation and senescence: a mini-review. Gerontology. 2015;61(2):131-8.
21. Atamna H, Atamna W, Al-Eyd G, Shanower G, Dhahbi JM. Combined activation of the energy and cellular-defense pathways may explain the potent anti-senescence activity of methylene blue. Redox Biol. 2015 Dec;6:426-35. doi: 10.1016/j.redox.2015.09.004.
22. Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011 Jan 28;331(6016):456-61.
23. Zhang ZS, Wang J, Shen YB, et al. Dihydroartemisinin increases temozolomide efficacy in glioma cells by inducing autophagy. Oncol Lett. 2015 Jul;10(1):379-383.
24. Ohrt C, Li Q, Obaldia N, Im-Erbsin R, Xie L, Berman J. Efficacy of intravenous methylene blue, intravenous artesunate, and their combination in preclinical models of malaria. Malar J. 2014 Oct 21;13:415.
25. Olagnier D, Lavergne RA, Meunier E, et al. Nrf2, a PPARγ alternative pathway to promote CD36 expression on inflammatory macrophages: implication for malaria. PLoS Pathog. 2011 Sep;7(9):e1002254.
26. Serghides L, Kain KC. Mechanism of protection induced by vitamin A in falciparum malaria. Lancet. 2002 Apr 20;359(9315):1404-6.
27. Alzoubi K, Calabrò S, Bissinger R, Abed M, Faggio C, Lang F. Stimulation of suicidal erythrocyte death by artesunate. Cell Physiol Biochem. 2014;34(6):2232-44.
28. Föller M, Bobbala D, Koka S, Huber SM, Gulbins E, Lang F. Suicide for survival--death of infected erythrocytes as a host mechanism to survive malaria. Cell Physiol Biochem. 2009;24(3-4):133-40.
29. Alzoubi K, Calabrò S, Faggio C, Lang F. Stimulation of suicidal erythrocyte death by sulforaphane. Basic Clin Pharmacol Toxicol. 2015 Mar;116(3):229-35.
No comments:
Post a Comment